
CeO2 catalysed conversion of CO, NO2 and NO from first principles energetics
Abstract
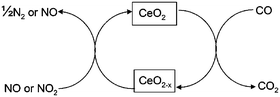
First principles calculations using density functional theory with corrections for on-site Coulomb interactions (DFT + U) are presented in which we compute the energy for the conversion of CO to CO2, NO2 to NO and NO to N2 over ceria surfaces. The surface sensitivity is discussed on the basis of the vacancy formation energies.
 Please wait while we load your content...
Please wait while we load your content...

