
Accurate ab initio determination of spectroscopic and thermochemical properties of mono- and dichlorocarbenes†
Abstract
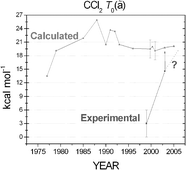
The best technically feasible values for the triplet–singlet energy gap and the enthalpies of formation of the HCCl and CCl2 radicals have been determined through the focal-point approach. The electronic structure computations were based on high-level coupled cluster (CC) methods, up to quadruple excitations (CCSDTQ), and large-size correlation-consistent basis sets, ranging from aug-cc-pVDZ to aug-cc-pV6Z, followed by extrapolation to the complete basis set limit. Small corrections due to core correlation, relativistic effects, diagonal Born–Oppenheimer correction, as well as harmonic and anharmonic zero-point vibrational energy corrections have been taken into account. The final estimates for the triplet–singlet energy gap, T0(ã), are 2170 ± 40 cm−1 for HCCl and 7045 ± 60 cm−1 for CCl2, favoring the singlet states in both cases. Complete quartic force fields in internal coordinates have been computed for both the ![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) and ã states of both radicals at the frozen-core CCSD(T)/aug-cc-pVQZ level. Using these force fields vibrational energy levels of {HCCl, DCCl, CCl2} up to {6000, 5000, 7000} cm−1 were calculated both by second-order vibrational perturbation theory (VPT2) and variationally. These results, especially the variational ones, show excellent agreement with the experimentally determined energy levels. The enthalpies of formation of HCCl (1A′) and CCl2(1A1), at 0 K, are 76.28 ± 0.20 and 54.54 ± 0.20 kcal mol−1, respectively.
and ã states of both radicals at the frozen-core CCSD(T)/aug-cc-pVQZ level. Using these force fields vibrational energy levels of {HCCl, DCCl, CCl2} up to {6000, 5000, 7000} cm−1 were calculated both by second-order vibrational perturbation theory (VPT2) and variationally. These results, especially the variational ones, show excellent agreement with the experimentally determined energy levels. The enthalpies of formation of HCCl (1A′) and CCl2(1A1), at 0 K, are 76.28 ± 0.20 and 54.54 ± 0.20 kcal mol−1, respectively.
 Please wait while we load your content...
Please wait while we load your content...

