
VRAI-selectivity: calculation of selectivity beyond transition state theory†
 a
and
Jonathan M.
Goodman
*a
a
and
Jonathan M.
Goodman
*a
Abstract
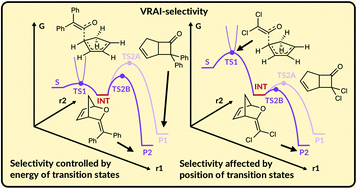
In recent years, a growing number of organic reactions in the literature have shown selectivity controlled by reaction dynamics rather than by transition state theory. Such reactions are difficult to analyse because the transition state theory approach often does not capture the subtlety of the energy landscapes the compounds traverse and, therefore, cannot accurately predict the selectivity. We present an algorithm that can predict the major product and selectivity for a wide range of potential energy surfaces where the product distribution is influenced by reaction dynamics. The method requires as input calculation of the transition states, the intermediate (if present) and the product geometries. The algorithm is quick and simple to run and, except for two reactions with long alkyl chains, calculates selectivity more accurately than transition state theory alone.
- This article is part of the themed collection: Mechanistic, computational & physical organic chemistry in OBC
 Please wait while we load your content...
Please wait while we load your content...

