
Exploring the charge localization and band gap opening of borophene: a first-principles study
 ab
Kun
Zhou,
*a
ab
Kun
Zhou,
*a
Abstract
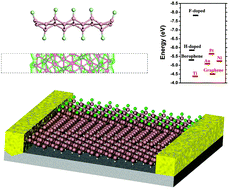
Recently synthesized two-dimensional (2D) boron, borophene, exhibits a novel metallic behavior rooted in the s–p orbital hybridization, distinctively different from other 2D materials such as sulfides/selenides and semi-metallic graphene. This unique feature of borophene implies new routes for charge delocalization and band gap opening. Herein, using first-principles calculations, we explore the routes to localize the carriers and open the band gap of borophene via chemical functionalization, ribbon construction, and defect engineering. The metallicity of borophene is found to be remarkably robust against H- and F-functionalization and the presence of vacancies. Interestingly, a strong odd–even oscillation of the electronic structure with width is revealed for H-functionalized borophene nanoribbons, while an ultra-high work function (∼7.83 eV) is found for the F-functionalized borophene due to its strong charge transfer to the atomic adsorbates.
 Please wait while we load your content...
Please wait while we load your content...

