
A bond-order potential for the Al–Cu–H ternary system†
 *a
*a
Abstract
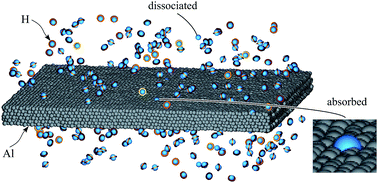
Al-Based Al–Cu alloys have a very high strength to density ratio, and are therefore important materials for transportation systems including vehicles and aircrafts. These alloys also appear to have a high resistance to hydrogen embrittlement, and as a result, are being explored for hydrogen related applications. To enable fundamental studies of mechanical behavior of Al–Cu alloys under hydrogen environments, we have developed an Al–Cu–H bond-order potential according to the formalism implemented in the molecular dynamics code LAMMPS. Our potential not only fits well to properties of a variety of elemental and compound configurations (with coordination varying from 1 to 12) including small clusters, bulk lattices, defects, and surfaces, but also passes stringent molecular dynamics simulation tests that sample chaotic configurations. Careful studies verified that this Al–Cu–H potential predicts structural property trends close to experimental results and quantum-mechanical calculations; in addition, it properly captures Al–Cu, Al–H, and Cu–H phase diagrams and enables simulations of H2 dissociation, chemisorption, and absorption on Al–Cu surfaces.
 Please wait while we load your content...
Please wait while we load your content...

