
A density functional study on the electronic structure, nature of bonding and reactivity of NO adsorbing Rh 0/±n (n = 2–8) clusters

Abstract
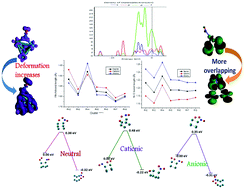
Systematic investigations on lowest energy NO adsorbing neutral and ionic Rhn (n = 2–8) clusters in the gas phase are executed with an all electron relativistic method using density functional theory (DFT) within the generalized gradient approximation. Geometrical parameters like bond length, adsorption energy, and vibrational stretching frequency and reactivity parameters such as electron density, density of states (DOS), LUMO (lowest unoccupied molecular orbital), HOMO (highest occupied molecular orbital), etc. are evaluated to understand the bonding nature as well as the binding interaction of NO with stable Rh0/±n (n = 2–8) clusters. RhnNO− clusters are found to be more stable on the basis of adsorption energy when compared to RhnNO0/+. Synergic bond formation is noticed between the rhodium atom and the NO molecule due to back-donation of electrons from the metal d-orbitals to the π* orbital of NO in the case of RhnNO0/−. Deformed electron density also suggests a stronger bond between the rhodium and N atoms of the NO molecule in the anionic adducts. HOMO and LUMO isosurface diagrams reveal that electrons are delocalized from the d-orbitals of rhodium (HOMO) to the π* orbital of NO (LUMO). Mülliken charge analysis along with electron density distribution shows the higher stability of RhnNO− clusters than that of RhnNO0/+. Energy profile diagrams for the conversion of NO to NO2 catalysed by neutral and ionic Rhn clusters reveal a lower activation barrier for the anionic rhodium clusters in comparison to Rh0/+n except for Rh7. Anionic Rh4 and Rh6 clusters are seen to be catalytically more active for the conversion of NO to NO2.
 Please wait while we load your content...
Please wait while we load your content...

