
COSMO-CAMPD: a framework for integrated design of molecules and processes based on COSMO-RS†
 a
L.
Fleitmann,
a
J.
Thien,
a
C.
Redepenning,
b
K.
Leonhard,
a
W.
Marquardt
b
and
A.
Bardow
*ac
a
L.
Fleitmann,
a
J.
Thien,
a
C.
Redepenning,
b
K.
Leonhard,
a
W.
Marquardt
b
and
A.
Bardow
*ac
Abstract
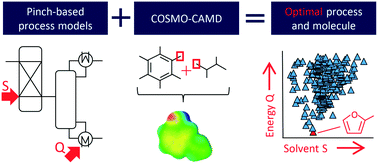
Molecules are often the key for energetically and economically efficient processes and thus need to be carefully selected. The simultaneous optimization of molecules and processes is addressed by computer-aided molecular and process design (CAMPD). Thereby, the design problem captures directly the complex interaction of molecules and processes. The success of CAMPD strongly relies on the available model to predict the molecular properties. Typically, simplified thermodynamic prediction methods such as group contribution (GC) methods are used. GC methods assume the additivity of functional groups and require interaction parameters determined in experiments which limits the molecular design space. In this work, we overcome these limitations by presenting COSMO-CAMPD, a framework for integrated design of molecules and processes based on COSMO-RS. COSMO-RS employs quantum-mechanics (QM)-based thermodynamic property predictions and thus does not rely on experimental data or group additivity. We embed the COSMO-RS predictions in an efficient QM calculation strategy using two COSMO-RS accuracy levels. This strategy reduces computational cost to less than 1 minute per molecule. Designed molecules are evaluated at the process level using highly efficient and thermodynamically accurate pinch-based process models. With the results from pinch-based process models, rigorous process flowsheet simulations are initialized and easily converged in a subsequent refinement step using Aspen Plus. We apply the COSMO-CAMPD framework to a case study for solvent design in a hybrid extraction–distillation process for the bio-based platform chemical γ-valerolactone. COSMO-CAMPD successfully designs molecules as solvents for an optimized process which reduces the energy demand by 63% in comparison to published literature solvents. The predictions of COSMO-CAMPD are challenged by liquid–liquid equilibrium experiments for the top designed commercially available solvents 2-methylfuran and toluene. For the most promising commercially available solvent with practical relevance, 2-methylfuran, the experiments lead to 50% reduction in process energy demand compared to the benchmark solvent n-butyl acetate corresponding well to the reduction of 47% predicted by COSMO-CAMPD. The experimental validation highlights the practical value of COSMO-CAMPD.
 Please wait while we load your content...
Please wait while we load your content...

