
Electrochemical behavior of a Rh(pentamethylcyclopentadienyl) complex bearing an NAD+/NADH-functionalized ligand†
 c
Koji
Tanaka
*c
c
Koji
Tanaka
*c
Abstract
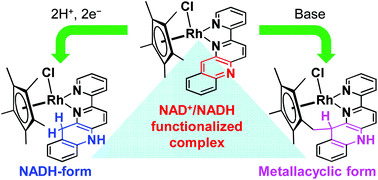
A RhCp* (Cp* = pentamethylcyclopentadienyl) complex bearing an NAD+/NADH-functionalized ligand, [RhCp*(pbn)Cl]Cl ([1]Cl, pbn = (2-(2-pyridyl)benzo[b]-1,5-naphthyridine)), was synthesized. The cyclic voltammogram of [1]Cl in CH3CN shows two reversible redox waves at E1/2 = −0.58 and −1.53 V (vs. the saturated calomel electrode (SCE)), which correspond to the RhIII/RhI and pbn/pbn˙− redox couples, respectively. The addition of acetic acid to the solution afforded the proton-coupled two-electron reduction of [1]Cl at −0.62 V, from which [RhCp*(pbnHH)Cl]+ was selectively generated, probably via a hydride transfer from a RhIII–hydride intermediate to the pbn ligand. Complex [1]Cl is stable under acidic conditions, whereas a methyl proton of the Cp* moiety dissociates under basic conditions. The resulting anionic methylene group attacks the para carbon of the free pyridine of pbn, accompanied by protonation of the nitrogen atom of the ligand. As a result, treatment of [1]Cl with a base produces selectively the cyclic complex [1CH]Cl, which bears a reduced pbn framework (pbnCH). [1CH]Cl forms 1 : 1 adducts with PhCOO−via hydrogen bonding. A similar adduct, formed by a Ru–pbnHH scaffold and RCOO− (R = CH3, C6H5), has been reported to react with CO2 to produce HCOO− under concomitant regeneration of Ru–pbn. The adduct of [1CH]Cl with PhCOO−, however, lacks such hydride-donor ability, due to a steric barrier in the molecular structure of [1CH]Cl, which hampers the hydride transfer.
 Please wait while we load your content...
Please wait while we load your content...

