
Membrane potential and dynamics in a ternary lipid mixture: insights from molecular dynamics simulations†

Abstract
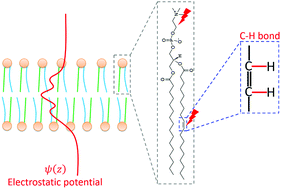
Transmembrane potential (Vm) plays critical roles in cell signaling and other functions. However, the impact of Vm on the structure and dynamics of membrane lipids and proteins, which are critical for the regulation of signaling, is still an open question. All-atom molecular dynamics (MD) simulation is emerging as a useful technique to address this issue. Previous atomistic MD simulations of pure or binary model membranes indicated that both ion imbalance and electric field can be used to generate Vm, but both approaches failed to yield structural changes in lipids with statistical significance. We hypothesized that a possible reason for this could be oversimplified membrane composition or limited sampling. In this work, we tested if and how Vm modulates the structure and dynamics of lipids in a physiologically relevant model membrane. Using a detailed side-by-side comparison, we first show that while both ion imbalance and electric field generate Vm in our complex membranes, only the latter could produce physiologically relevant Vm. We further show that double bonds in lipid acyl chains have a relatively large sensitivity to Vm. A single-bilayer model with an electric field showed the highest sensitivity in simulations under the isothermal–isobaric (NPT) ensemble, reproducing expected responses of head-group dipoles to Vm and suggesting that this approach may be more suitable for studying the structural effects of Vm. Our findings also shed light on the relationship between the macroscopic Vm and its atomic-level underpinnings.
 Please wait while we load your content...
Please wait while we load your content...

