
Theoretical insight into the vibrational spectra of metal–water interfaces from density functional theory based molecular dynamics†
 *ac
*ac
Abstract
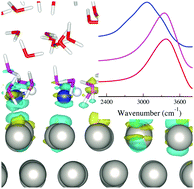
Understanding the structures of electrochemical interfaces at the atomic level is key to developing efficient electrochemical cells for energy storage and conversion. Spectroscopic techniques have been widely used to investigate the structures and vibrational properties of the interfaces. The interpretation of these spectra is however not straightforward. In this work, density functional theory based molecular dynamics simulations were performed to study the vibrational properties of the Pt(111)– and Au(111)–water interfaces. It was found that the specific adsorption of some surface water on Pt(111) leads to a partial charge transfer to the metal, and strong hydrogen bonding with neighbouring water molecules, which resolves the interpretation of the elusive O–H stretching peak at around 3000 cm−1 observed in some experiments.
 Please wait while we load your content...
Please wait while we load your content...

