
Li deposition and desolvation with electron transfer at a silicon/propylene-carbonate interface: transition-state and free-energy profiles by large-scale first-principles molecular dynamics†

Abstract
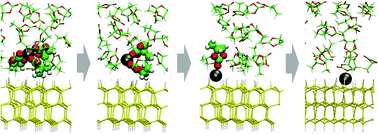
We report the result of a large-scale first-principles molecular dynamics simulation under different electric biases performed to understand the charge transfer process coupling with lithium deposition and desolvation processes. We applied the effective screening medium (ESM) method to control the bias across the electrode/solution interface, and simulated a series of Li de-solvation and Li-deposition reactions occurring under the bias. Solvated Li+ in the bulk propylene carbonate migrates to the Si electrode surface and gradually de-solvates through the transition state. Introducing the blue-moon ensemble method, we determined the possible structures and activation energies for the transition states.
 Please wait while we load your content...
Please wait while we load your content...

