
Modelling molecular adsorption on charged or polarized surfaces: a critical flaw in common approaches†
Kristof M.
Bal  *a
and
Erik C.
Neyts
a
*a
and
Erik C.
Neyts
a
*a
and
Erik C.
Neyts
a
Abstract
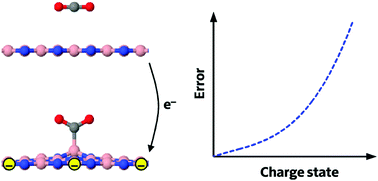
A number of recent computational material design studies based on density functional theory (DFT) calculations have put forward a new class of materials with electrically switchable chemical characteristics that can be exploited in the development of tunable gas storage and electrocatalytic applications. We find systematic flaws in almost every computational study of gas adsorption on polarized or charged surfaces, stemming from an improper and unreproducible treatment of periodicity, leading to very large errors of up to 3 eV in some cases. Two simple corrective procedures that lead to consistent results are proposed, constituting a crucial course correction to the research in the field.
 Please wait while we load your content...
Please wait while we load your content...

