
Density functional theory studies on the solvent effects in Al(H2O)63+ water-exchange reactions: the number and arrangement of outer-sphere water molecules†

Abstract
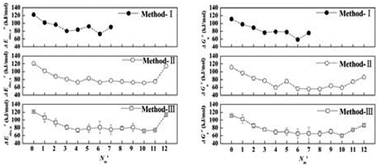
Density functional theory (DFT) calculations combined with cluster models are performed at the B3LYP/6-311+G(d,p) level for investigating the solvent effects in Al(H2O)63+ water-exchange reactions. A “One-by-one” method is proposed to obtain the most representative number and arrangement of explicit H2Os in the second hydration sphere. First, all the possible ways to locate one explicit H2O in second sphere (Nm′ = 1) based on the gas phase structure (Nm′ = 0) are examined, and the optimal pathway (with the lowest energy barrier) for Nm′ = 1 is determined. Next, more explicit H2Os are added one by one until the inner-sphere is fully hydrogen bonded. Finally, the optimal pathways with Nm′ = 0–7 are obtained. The structural and energetic parameters as well as the lifetimes of the transition states are compared with the results obtained with the “Independent-minimum” method and the “Independent-average” method, and all three methods show that the pathway with Nm′ = 6 may be representative. Our results give a new idea for finding the representative pathway for water-exchange reactions in other hydrated metal ion systems.
 Please wait while we load your content...
Please wait while we load your content...

