
Molecular dynamics of the halloysite nanotubes

Abstract
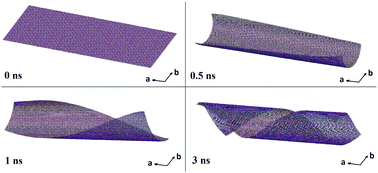
We report large-scale and long-time molecular dynamics simulations demonstrating the transformation of a single kaolin alumosilicate sheet to a halloysite nanotube. The models we consider contain up to 5 × 105 atoms, which is two orders of magnitude larger than that used in previous theoretical works. It was found that the temperature plays a crucial role in the formation of the rolled geometry of the halloysite. For the models with periodic boundary conditions, we observe the tendency to form twin-tube structures, which is confirmed experimentally by atomic force microscopy imaging. The molecular dynamics calculations show that the rate of the rolling process is very sensitive to the choice of the winding axis and varies from 5 ns to 25 ns. The effects of the open boundary conditions and the initial form of the kaolin alumosilicate sheet are discussed. Our simulation results are consistent with experimental TEM and AFM halloysite tube imaging.
 Please wait while we load your content...
Please wait while we load your content...

