
Exploiting coordination geometry to selectively predict the σ-donor and π-acceptor abilities of ligands: a back-and-forth journey between electronic properties and spectroscopy†

Abstract
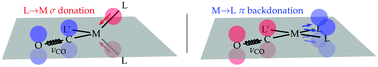
Through an analysis of eighty tetrahedral and square-planar metal carbonyls of general formula [M(CO)(L′)(L)2] including newly synthesized chlorocarbonyl rhodium complexes with chelating atropoisomeric diphosphanes, we show how coordination geometry can switch the carbonyl stretching frequency into a selective probe of the σ-donor and π-acceptor abilities of the ligands. We thus provide a framework whereby the σ-donation and π-backdonation constituents of the Dewar–Chatt–Duncanson model can be quantitatively predicted through spectroscopic data on coordinated CO moieties and vice versa.
 Please wait while we load your content...
Please wait while we load your content...

