
Predicted energy–structure–function maps for the evaluation of small molecule organic semiconductors†

Abstract
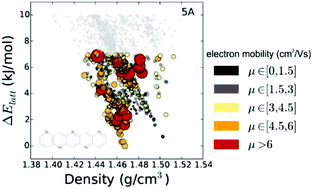
The computational assessment of materials through the prediction of molecular and crystal properties could accelerate the discovery of novel materials. Here, we present calculated energy–structure–function maps based on crystal structure prediction for a series of hypothetical organic molecular semiconductors, to demonstrate their utility in evaluating molecules prior to their synthesis. Charge transfer in organic semiconductors relies on the degree of π-conjugation and overlap of the π-systems of neighbouring molecules in the solid state. We explore the effects of varying levels of nitrogen substitution on the crystal packing and charge transport properties of aza-substituted pentacenes, in which C–H⋯N hydrogen bonding is predicted to favour co-planar molecular packing in preference to the edge-to-face herringbone packing seen for pentacene. The charge mobilities of predicted structures in the energy range of expected polymorphism were calculated, highlighting the important balance between intra- and intermolecular properties when designing novel organic semiconductors. The use of predicted landscapes to rank molecules according to their likely properties is discussed.
 Please wait while we load your content...
Please wait while we load your content...

