
Structures, mobility and electronic properties of point defects in arsenene, antimonene and an antimony arsenide alloy
 *a
and
Youyong
Li
*a
*a
and
Youyong
Li
*a
Abstract
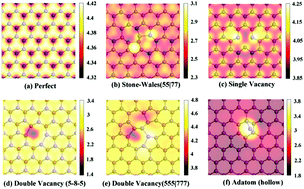
Defects are unavoidable during the synthesis of materials, especially for two-dimensional (2D) nanomaterials. They are usually seen as detrimental to device properties, but sometimes bring about new beneficial effects. In order to clarify the influence of defects on the structural and electronic properties, we have performed first-principles calculations to systematically investigate the structural stability, mobility and electronic properties of typical point defects in 2D arsenene (h-As), antimonene (h-Sb) and antimony arsenide (h-AsSb), including the Stone–Wales defects, single vacancies (SVs), double vacancies (DVs) and adatoms. To provide visual guidance for experimental observations, scanning tunnelling microscopy (STM) images are simulated. Compared to defects in graphene and silicene, these defects form more easily with lower formation energies, and SVs can diffuse very quickly to the edges with a lower diffusion barrier of less than 1 eV. Monolayer arsenene, antimonene and antimony arsenide are indirect band gap semiconductors, and the defective structures significantly reduce the band gaps. Most of the SV and adatom defects carry magnetic moments due to the dangling bonds resulting from the absent or extra atom. Our present results have demonstrated that the point defects induce significant effects on the electronic properties of pristine arsenene, antimonene and the antimony arsenide alloy, which should be considered in their future applications.
 Please wait while we load your content...
Please wait while we load your content...

