
Computational investigation of CO2 electroreduction on tin oxide and predictions of Ti, V, Nb and Zr dopants for improved catalysis†

Abstract
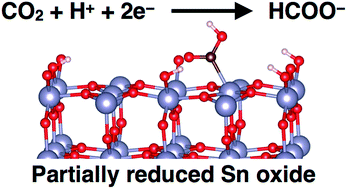
We have used computational quantum chemistry and atomistic thermodynamics to identify reaction intermediates for CO2 electroreduction on partially reduced tin oxide electrodes. We find that a variety of different surface morphologies are thermodynamically accessible under reducing potentials with adsorbed CO2, adsorbed H atoms, and O vacancy defects that represent partially reduced states. Our work supports prior conclusions from experimental findings that the active catalyst for this system is a hydroxylated and partially reduced SnO2 surface that forms under operating conditions for CO2 reduction. Employing thermodynamic descriptors and the computational hydrogen electrode model, we predict that doping Sn electrodes with Ti, V, Nb, or Zr will result in lower overpotentials for CO2 reduction compared to undoped tin oxide.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

