
Predicting electronic structure properties of transition metal complexes with neural networks†
 a
and
Heather J.
Kulik
*a
a
and
Heather J.
Kulik
*a
Abstract
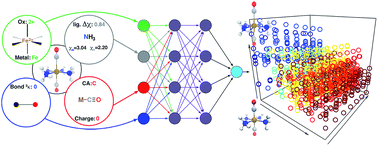
High-throughput computational screening has emerged as a critical component of materials discovery. Direct density functional theory (DFT) simulation of inorganic materials and molecular transition metal complexes is often used to describe subtle trends in inorganic bonding and spin-state ordering, but these calculations are computationally costly and properties are sensitive to the exchange–correlation functional employed. To begin to overcome these challenges, we trained artificial neural networks (ANNs) to predict quantum-mechanically-derived properties, including spin-state ordering, sensitivity to Hartree–Fock exchange, and spin-state specific bond lengths in transition metal complexes. Our ANN is trained on a small set of inorganic-chemistry-appropriate empirical inputs that are both maximally transferable and do not require precise three-dimensional structural information for prediction. Using these descriptors, our ANN predicts spin-state splittings of single-site transition metal complexes (i.e., Cr–Ni) at arbitrary amounts of Hartree–Fock exchange to within 3 kcal mol−1 accuracy of DFT calculations. Our exchange-sensitivity ANN enables improved predictions on a diverse test set of experimentally-characterized transition metal complexes by extrapolation from semi-local DFT to hybrid DFT. The ANN also outperforms other machine learning models (i.e., support vector regression and kernel ridge regression), demonstrating particularly improved performance in transferability, as measured by prediction errors on the diverse test set. We establish the value of new uncertainty quantification tools to estimate ANN prediction uncertainty in computational chemistry, and we provide additional heuristics for identification of when a compound of interest is likely to be poorly predicted by the ANN. The ANNs developed in this work provide a strategy for screening transition metal complexes both with direct ANN prediction and with improved structure generation for validation with first principles simulation.
 Please wait while we load your content...
Please wait while we load your content...

