
Generalized-active-space pair-density functional theory: an efficient method to study large, strongly correlated, conjugated systems†

Abstract
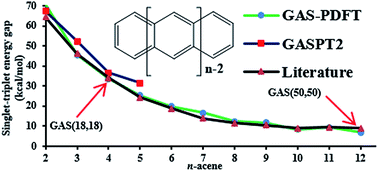
Predicting ground- and excited-state properties of open-shell organic molecules by electronic structure theory can be challenging because an accurate treatment has to correctly describe both static and dynamic electron correlation. Strongly correlated systems, i.e., systems with near-degeneracy correlation effects, are particularly troublesome. Multiconfigurational wave function methods based on an active space are adequate in principle, but it is impractical to capture most of the dynamic correlation in these methods for systems characterized by many active electrons. We recently developed a new method called multiconfiguration pair-density functional theory (MC-PDFT), that combines the advantages of wave function theory and density functional theory to provide a more practical treatment of strongly correlated systems. Here we present calculations of the singlet–triplet gaps in oligoacenes ranging from naphthalene to dodecacene. Calculations were performed for unprecedently large orbitally optimized active spaces of 50 electrons in 50 orbitals, and we test a range of active spaces and active space partitions, including four kinds of frontier orbital partitions. We show that MC-PDFT can predict the singlet–triplet splittings for oligoacenes consistent with the best available and much more expensive methods, and indeed MC-PDFT may constitute the benchmark against which those other models should be compared, given the absence of experimental data.
- This article is part of the themed collection: Celebrating Excellence in Research: 100 Women of Chemistry
 Please wait while we load your content...
Please wait while we load your content...

