
Donor–acceptor type A2B2 porphyrins: synthesis, energy transfer, computational and electrochemical studies†
Abstract
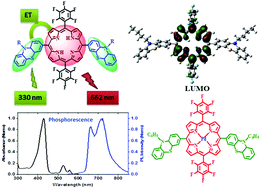
A series of donor–acceptor type trans-A2B2 porphyrins and their Zn(II) and Pd(II) complexes 5–13 have been synthesized and characterized by various spectroscopic techniques. The effect of the donor moieties (e.g., N-butylcarbazole, N-butylphenothiazine, and triphenylamine) on the spectroscopic properties of the porphyrins has been studied. The structural changes indeed affected the optical and electrochemical properties of these porphyrins. Higher energy shifts of the Soret bands were observed for porphyrins upon varying the donor moieties. The electrochemical studies of all the derivatives indicated increased interactions between the donor groups and the porphyrin core, which in turn are reflected in the anodic shifts in their reduction potentials. Both steady-state and time-resolved fluorescence studies revealed effective energy transfer (EET; up to 87%) from donor groups to the porphyrin core in the porphyrins, 5–10. The palladium(II) porphyrin complexes, 11–13, showed characteristic phosphorescence in the near IR region. Density functional theory (DFT) studies support the presence of donor–acceptor interaction between the porphyrin core and the meso-substituents in the dyads. Density functional theory (DFT) and time dependent-density functional theory (TD-DFT) studies showed that in 5, 8 and 11, the transitions are of π → π* type; whereas in the other molecules viz.6, 7, 9, 10, 12 and 13 intramolecular charge transfer (ICT) is involved in all the respective highest intensity absorption transitions.
 Please wait while we load your content...
Please wait while we load your content...

