
Rearrangement of vinyl allene oxide geometric isomers to cyclopentenones. Further computational insights with biologically relevant model systems†

Abstract
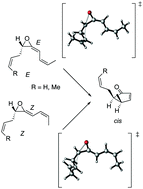
Pathways for the rearrangement of the E and Z isomers of allyl- and methyl-substituted vinyl allene oxides to stereodefined cyclopentenones have been studied by DFT computations. Regardless of the reactant geometry, cis-configured cyclopentenones are found to be formed in a stepwise cascade comprising as key steps the ring opening of the oxirane to give an oxidopentadienyl diradical, its isomerization, and electrocyclization. An allyl substituent at the Csp3 atom of the starting vinyl allene oxide induces opposite effects on the activation energies for ring opening: a decrease owing to assistance by homoconjugation for the out motion and an increase due to the stereoelectronic stabilization of the reactant. As a result, allyl- and methyl-substituted vinyl allene oxides exhibit comparable activation energies. Only model systems with crotyl substituents afford lower activation energies than the methyl counterparts due to the additional stabilization of the forming charge deficiency at a secondary carbon by homoconjugation. Moreover, upon homoconjugative interaction reactants of Z geometry are predicted to undergo cyclization more readily than the E isomers. The results with Z-crotyl substituent are congruent with the spontaneous rearrangement of natural vinyl allene oxide derived from α-linolenic acid to a racemic cis-cyclopentenone (12-oxo-PDA).
 Please wait while we load your content...
Please wait while we load your content...

