
The local electronic properties of individual Pt atoms adsorbed on TiO2(110) studied by Kelvin probe force microscopy and first-principles simulations†
 *abc
*abc
Abstract
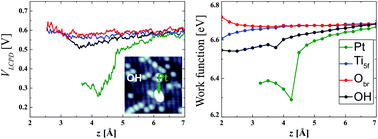
Noble metal nanostructures dispersed on metal oxide surfaces have applications in diverse areas such as catalysis, chemical sensing, and energy harvesting. Their reactivity, chemical selectivity, stability, and light absorption properties are controlled by the interactions at the metal/oxide interface. Single-atom metal adsorbates on the rutile TiO2(110)-(1 × 1) surface have become a paradigmatic model to characterize those interactions and to understand the unique electronic properties of these supported nanostructures. We combine Kelvin probe force microscopy (KPFM) experiments and density functional theory (DFT) calculations to investigate the atomic-scale variations in the contact potential difference of individual Pt atoms adsorbed on a hydroxylated (h) TiO2(110)-(1 × 1) surface. Our experiments show a significant drop in the local contact potential difference (LCPD) over Pt atoms with respect to the TiO2 surface, supporting the presence of an electron transfer from the Pt adsorbates to the substrate. We have identified two characteristic regimes by LCPD spectroscopy. At far tip–sample distances, LCPD values show a weak distance dependence and can be attributed to the intrinsic charge transfer from Pt to the oxide support. Beyond the onset of short-range chemical interactions, LCPD values exhibit a strong distance dependence that we ascribe to the local structural and charge rearrangements induced by the tip–sample interaction. These findings also apply to other electropositive adsorbates such as potassium and the hydrogen atoms forming the OH groups that are present on the h-TiO2(110) surface, promoting KPFM as a suitable tool for the understanding of electron transfer in catalytically active materials.
 Please wait while we load your content...
Please wait while we load your content...

