
A detailed investigation into the geometric and electronic structures of CoB Qn (n = 2–10, Q = 0, −1) clusters
 *bc
*bc
Abstract
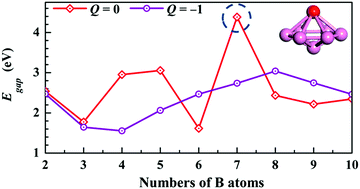
Here, transition metal (TM) cobalt-centered pure boron clusters were examined using both the CALYPSO (Crystal structure AnaLYsis by Particle Swarm Optimization) method and DFT (density functional theory) calculations. The ground states of CoBQn (n = 2–10, Q = 0, −1) mainly featured planar and quasi-planar structures except for neutral CoB7 which had an umbrella structure. Furthermore, analysis of the stability and electronic properties showed that the magic CoB7 cluster possessed great stability and a surprisingly large HOMO–LUMO gap. The simulated photoelectron spectra (PES) of the structures of the anionic ground state clusters are in agreement with the experimental spectra, indicating that the obtained ground state structures are the true minima. Molecular orbital (MO) and adaptive natural density partitioning (AdNDP) analyses revealed that the 3d atomic orbital (AO) of the Co atom and most of the B–B σ-bonds in the B6 rings contributed to the stabilization of the neutral, umbrella-shaped, magic CoB7 cluster.
 Please wait while we load your content...
Please wait while we load your content...

