
Vibrational and conformational studies of 1,3-diaminopropane and its N-deuterated and N-ionised derivatives†

Abstract
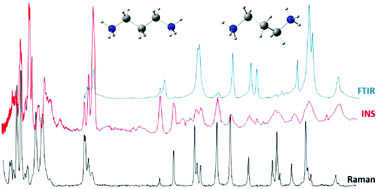
A vibrational and conformational analysis of the linear alkylpolyamine 1,3-diaminopropane (1,3-dap) is reported, using vibrational spectroscopy (Raman, Fourier Transform Infrared (FTIR) and inelastic neutron scattering (INS)) coupled to theoretical approaches at the Density Functional Theory (DFT) level. The quantum mechanical calculations were carried out using the mPW1PW functional and the 6-31G* basis set, for the isolated molecule, the condensed phase, and solutions in both water and carbon tetrachloride. The most stable geometries were calculated to be GGG′G and TG′GG′ for the gaseous phase and the CCl4 solution, and TTTT, TGTT and TTTG for the condensed phase and the aqueous solution. Since the relative populations obtained for the different 1,3-dap conformers were very similar, the corresponding experimental spectra reflect the presence of a mixture of species. The vibrational data obtained for 1,3-dap in its pure form – unprotonated, totally protonated (N-ionised) and N-deuterated – as well as for its aqueous and CCl4 solutions, were assigned in the light of the theoretical results presently obtained and experimental data previously gathered for similar compounds.
 Please wait while we load your content...
Please wait while we load your content...

