
B-Hb⋯π interactions in benzene–borazine sandwich and multidecker complexes: a DFT study†

Abstract
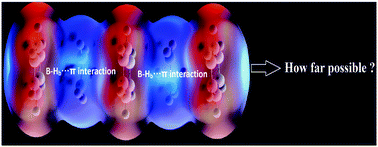
The stabilization energy of benzene–borazine sandwich and multidecker complexes bridged via B2H6 have been estimated using density functional theory. The gas and solvent phase calculations showed that the stabilization energy increases with the size of the complexes; in contrast, trifling reduction in the average stabilization energy is observed with increasing size of the complexes. The values of reactivity parameters, namely the energy of the HOMO, global hardness and chemical potential, suggest that the benzene complexes are more stable as compared to the borazine complexes. The complexes with as many as six aromatic rings containing B-Hb⋯π interactions are studied and are predicted to be stable (in terms of stabilization energy). The effect of solvents on the stability of the complexes is not so pronounced.
 Please wait while we load your content...
Please wait while we load your content...

