
Predicting protein lysine phosphoglycerylation sites by hybridizing many sequence based features†
 *a
*a
Abstract
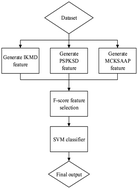
Post-translational modification (PTM) is essential for many biological processes. Covalent and generally enzymatic modification of proteins can impact the activity of proteins. Modified proteins would have more complex structures and functions. Knowing whether a specific residue is modified or not is significant to unravel the function and structure of this protein. As experimental approaches to discover protein PTM sites are always costly and time consuming, computational prediction methods are desirable alternative methods. Lysine phosphoglycerylation is a type of newly discovered PTM that is related to glycolytic process and glucose metabolism. Since the lysine phosphoglycerylation process requires no catalytic enzyme, its site selectivity mechanism is still not fully understood. In this study, we designed a novel computational method, namely PhoglyPred, to identify lysine phosphoglycerylation sites. By utilizing several different protein sequence descriptors, PhoglyPred achieved an overall accuracy of 90.3% in a Jackknife test, which is better than other state-of-the-art predictors. By analyzing the importance of different features using the F-score, we found several important sequence features, which may benefit future studies in understanding the site selectivity mechanism of lysine phosphoglycerylation.
 Please wait while we load your content...
Please wait while we load your content...