
Understanding the microscopic binding mechanism of hydroxylated and sulfated polybrominated diphenyl ethers with transthyretin by molecular docking, molecular dynamics simulations and binding free energy calculations†
 ab
ab
Abstract
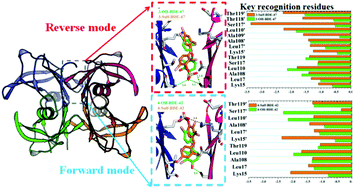
Polybrominated diphenyl ethers (PBDEs), one typical type of persistent environmental contaminant, have toxicological effects such as disrupting thyroid homeostasis in the human body. The high binding affinities of hydroxylated metabolites of PBDEs (OH-PBDEs) with transthyretin (TTR) were considered to be one major reason for their extraordinary capacity of passing through the blood–brain barrier via competitive thyroid hormone (T4) transport protein binding. Recent findings showed that sulfated PBDEs can be formed in human liver cytosol as phase-II metabolites. However, experimentally determined data for the TTR binding potential of the sulfated PBDEs are still not available. Therefore, molecular docking and molecular dynamics (MD) simulations were employed in the present study to probe the molecular basis of TTR interacting with hydroxylated and sulfated PBDEs at the atomic level. The docking scores of LeDock were used to construct the structure-based predictive model. The calculated results showed that the sulfated PBDEs have stronger affinity for TTR than the corresponding OH-PBDEs. Further analysis of structural characteristics based on MD simulations indicated that upon the binding of PBDE metabolites, the stability of TTR was enhanced and the dissociation rate of the tetrameric protein structure was potentially decreased. Subsequent binding free energy calculations implied that van der Waals interactions are the dominant forces for the binding of these metabolites of PBDEs at the T4 site of TTR. The residues Ser117/Ser117′ and Lys15/Lys15′ were identified, by both residue energy decomposition and computational alanine-scanning mutagenesis methods, as key residues which play an important role in determining the binding orientations of the –OSO3− group of sulfated PBDEs by formation of either hydrogen bonds or electrostatic interactions, respectively. In general, the combination of docking calculations with MD simulations provided a theoretically toxicological assessment for the metabolites of PBDEs, deep insight into the recognition mechanism of TTR for these compounds, and thus more comprehensive understanding of the thyroid-related toxic effects of PBDEs as well.
 Please wait while we load your content...
Please wait while we load your content...