
Design principles from multiscale simulations to predict nanostructure in self-assembling ionic liquids†

Abstract
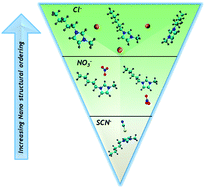
Molecular dynamics simulations (up to the nanoscale) were performed on the 3-methyl-1-pentylimidazolium ionic liquid cation paired with three anions; chloride, nitrate, and thiocyanate as aqueous mixtures, using the effective fragment potential (EFP) method, a computationally inexpensive way of modeling intermolecular interactions. The simulations provided insight (preferred geometries, radial distribution functions and theoretical proton NMR resonances) into the interactions within the ionic domain and are validated against 1H NMR spectroscopy and small- and wide-angle X-ray scattering experiments on 1-decyl-3-methylimidazolium. Ionic liquids containing thiocyanate typically resist gelation and form poorly ordered lamellar structures upon mixing with water. Conversely, chloride, a strongly coordinating anion, normally forms strong physical gels and produces well-ordered nanostructures adopting a variety of structural motifs over a very wide range of water compositions. Nitrate is intermediate in character, whereby upon dispersal in water it displays a range of viscosities and self-assembles into nanostructures with considerable variability in the fidelity of ordering and symmetry, as a function of water content in the binary mixtures. The observed changes in the macro and nanoscale characteristics were directly correlated to ionic domain structures and intermolecular interactions as theoretically predicted by the analysis of MD trajectories and calculated RDFs. Specifically, both chloride and nitrate are positioned in the plane of the cation. Anion to cation proximity is dependent on water content. Thiocyanate is more susceptible to water insertion into the second solvent shell. Experimental 1H NMR chemical shifts monitor the site-specific competition dependence with water content in the binary mixtures. Thiocyanate preferentially sits above and below the aromatic ring plane, a state disallowing interaction with the protons on the imidazolium ring.
- This article is part of the themed collection: Ionic liquids: from fundamental properties to practical applications
 Please wait while we load your content...
Please wait while we load your content...

