
Ferrate(vi) initiated oxidative degradation mechanisms clarified by DFT calculations: a case for sulfamethoxazole†
 *b
Yi
Luo
*a
*b
Yi
Luo
*a
Abstract
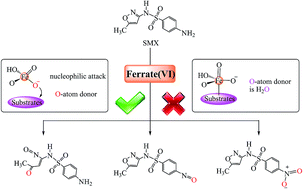
Ferrate(VI) is an efficient and environmentally friendly oxidant for the degradation of organic micropollutants. However, the related mechanism for the degradation is ambiguous and can hardly be elucidated empirically due to the rapid oxidation process and unstable intermediates for experimental trapping. Herein we performed density function theory (DFT) calculations to unveil the mechanism of ferrate(VI)-mediated degradation, taking sulfamethoxazole as a model compound. The results show that nucleophilic attack (rather than electrophilic attack) of HFeO4− on the isoxazole moiety of sulfamethoxazole initiates the subsequent degradations, and ferrate(VI) rather than the water molecule provides O atoms for the oxidation of the nitroso group and isoxazole moiety. Electron delocalization from the Fe atom to the isoxazole moiety is crucial for the ring-opening of isoxazole, and organometallic intermediates suggested previously are not the necessary ones in the oxidation of sulfamethoxazole by HFeO4−. Thus, this study has theoretically clarified the ferrate(VI) oxidation mechanisms for a representative sulfonamide, which were also partially corroborated by the intermediates and products observed in the previous experimental studies for phosphite and tryptophan. This study provides an exemplification on the application of quantum chemical calculations to clarify the degradation pathways of organic micropollutants, which is important for the prediction of degradation products needed in their engineering design.
- This article is part of the themed collection: QSARs and computational chemistry methods in environmental chemical sciences
 Please wait while we load your content...
Please wait while we load your content...

