
Stereospecific control of the metal-centred chirality of rhodium(iii) and iridium(iii) complexes bearing tetradentate CNN′P ligands†
 a
Ricardo
Rodríguez,
*a
Isabel
Méndez,
a
Vincenzo
Passarelli,
*ab
Fernando J.
Lahoz,
a
Pilar
García-Orduña
a
and
Daniel
Carmona
*a
a
Ricardo
Rodríguez,
*a
Isabel
Méndez,
a
Vincenzo
Passarelli,
*ab
Fernando J.
Lahoz,
a
Pilar
García-Orduña
a
and
Daniel
Carmona
*a
Abstract
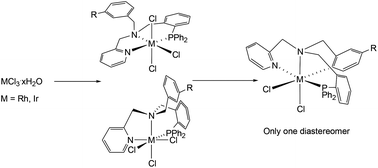
Ligands LH1–LH3 have been prepared by two successive condensation/reduction steps. These ligands react with MCl3 (M = Rh, Ir) rendering the trichlorido complexes [MCl3(κ3N,N′,P-LH)] (M = Rh, LH = LH1 (1), LH2 (2), LH3 (3); M = Ir, LH = LH1, (4)) as racemic mixtures of fac and mer isomers. Only one of the two possible fac isomers was detected. The mer isomer of the rhodium compounds 1–3 quantitatively isomerizes to the more stable fac isomer, whereas the mer isomer of the iridium complex 4 does not. DFT calculations indicate a dissociative pathway for this isomerization. In the presence of acetate or trifluoroacetate, complexes 1–3 or 4, respectively, undergo cyclometallation of their free benzylic arm affording the corresponding dichlorido compounds [MCl2(κ4C,N,N′,P-L)] (M = Rh, L = L1 (5), L2 (6), L3 (7); M = Ir, L = L1 (8)). Only one of the three possible enantiomeric pairs of coordination isomers was detected. The configuration at the stereogenic centres, namely the metal and the iminic nitrogen atom is stereospecifically predetermined. DFT calculations reveal that the cyclometallation follows an acetate-assisted mechanism and indicate that the isolated isomers are the most stable. Complexes 1–8 have been characterized by analytical and spectroscopic means and by the determination of the crystal structures of the complexes 1, 3 and 5–8 by X-ray diffractometry.
 Please wait while we load your content...
Please wait while we load your content...

