
A multiconfigurational approach to the electronic structure of trichromium extended metal atom chains†
 a
C.
de Graaf
*ac
a
C.
de Graaf
*ac
Abstract
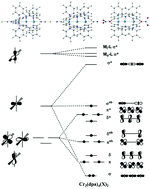
Density functional theory, Complete Active Space Self-Consistent Field (CASSCF) and perturbation theory (CASPT2) methodologies have been used to explore the electronic structure of a series of trichromium Extended Metal Atom Chains (EMACS) with different capping ligands. The study is motivated by the very different structural properties of these systems observed in X-ray experiments: the CN−-capped example has a symmetric Cr3 unit while for the NO3−-capped analogue the same unit has two very different Cr–Cr bond lengths. Density functional theory fails to locate an unsymmetric minimum for any of the systems studied, although the surface corresponding to the asymmetric stretch is very flat. CASPT2, in contrast, does locate an unsymmetric minimum only for the NO3−-capped system, although again the surface is very flat. We use the Generalized active space (GASSCF) technique and effective Hamiltonian theory to interrogate the multi-configurational wavefunctions of these systems, and show that the increase in the σ–σ* separation as the chain becomes unsymmetric plays a defining role in the stability of the ground state and its expansion in terms of configuration state functions.
 Please wait while we load your content...
Please wait while we load your content...

