
Complexes of MN2S2·Fe(η5-C5R5)(CO) as platform for exploring cooperative heterobimetallic effects in HER electrocatalysis†

Abstract
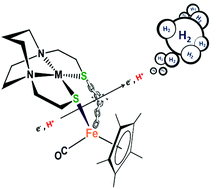
The control of aggregation at sulfur by metallodithiolates (MN2S2) has made them prime candidates as building blocks for the synthesis of biomimetics of various bimetallic enzyme active sites, with reactivity consequences implicating redox control by both metal centers. Recent studies of MN2S2 (M = Ni2+, Fe(NO)2+) bound to [(η5-C5H5)Fe(CO)]+ as electrocatalysts for proton reduction, the hydrogen evolution reaction, demonstrated reduction-induced hemi-lability of the bridging cis-dithiolates as a key step in the electrochemical proton reduction process (Ding, et al., J. Am. Chem. Soc., 2016, 138, 12920–12927). The MN2S2·Fe(η5-C5R5)(CO) platform offers numerous possibilities for tuning the electronic character of the M(μ-S2)Fe core. As well as modifying M within the metallodithiolate ligand, replacing H by CH3 at the η5-C5R5 moiety increases the electron density at the Fe center, which might facilitate the reductive Fe–S bond cleavage. Although release of a free thiolate in these hemi-labile ligands creates a needed internal pendant base, this benefit might be countered by the increase in over-potential for addition of the first electron. Herein we report the preparation and characterization of four bimetallic aggregates with the (η5-C5R5)Fe(CO) (R = H, CH3; Fe′ or Fe*′, respectively) or the dicarbonyl (η5-C5R5)Fe(CO)2 scaffold (R = H, CH3; Fe′′ or Fe*′′, respectively) bound to redox active MN2S2 ligands (M = Ni2+, Co(NO)2+; N2S2 = bismercaptoethane diazacycloheptane) Co-Fe*′, Ni-Fe*′, Co-Fe′ and Co-Fe*′′ complexes. The bidentate complexes were found to be electrocatalysts for proton reduction, although at high over-potential, especially for the derivatives of the electron-rich (η5-C5(CH3)5)Fe(CO)+. The turnover (TON) and turnover frequencies (TOF) were determined and found to be comparable to the previously reported MN2S2·Fe(η5-C5H5)(CO)+ analogues.
- This article is part of the themed collection: Multimetallic complexes: synthesis and applications
 Please wait while we load your content...
Please wait while we load your content...

