
Kinetics of hydrogen adsorption during catalytic reactions on transition metal surfaces

Abstract
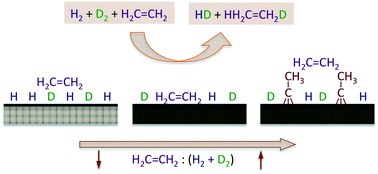
A study of the kinetics of the hydrogenation of ethylene promoted by hydrogen was performed by using a high-flux molecular beam. The experiments were designed to probe the intermediate pressure regime (around the mTorr range) associated with the transition between the surface-science research carried out under ultrahigh vacuum (UHV) environments and the catalytic studies performed under atmospheric conditions, the so-called “pressure gap” that is often mentioned but seldom addressed in catalytic work. In addition, the experiments were also focused on the characterization of the kinetics of both hydrogen isotope scrambling and ethylene hydrogenation simultaneously, by using H2 + D2 + C2H4 reaction mixtures and by following the time evolution of all of the products, HD and the different C2X6 isotopologues (X = H or D), in order to estimate the influence of the kinetics of the hydrogen uptake on the overall olefin conversion rates under reaction conditions. It was found that the addition of ethylene to the H2 + D2 beam leads to a significant decrease in the probability for HD production, but that the olefin hydrogenation reaction can still be sustained catalytically under the conditions of our experiments. Three kinetic regimes were identified with increasing partial pressure (or flux) of ethylene in the reaction mixture. The first, seen for mixtures with less than 10 parts per million of ethylene, shows a steady-state production of ethane with kinetics similar to those reported from UHV studies, with a rate law dependent linearly on both ethylene partial pressure and hydrogen atom coverage. The surface is mainly covered with hydrogen, and ethane formation occurs primarily via a previously unidentified “reverse” Eley–Rideal mechanism where olefin molecules from the gas phase pick up two hydrogen (deuterium) atoms upon impingement on the surface. A second, intermediate regime is seen for mixtures with up to about 1% of ethylene. In that case, HD production is still relatively fast, albeit the rate decreases slowly with increasing ethylene pressure, and the catalytic activity is mainly controlled by the coverage of the reversibly adsorbed ethylene, which partially blocks the hydrogen uptake (some di-σ ethylene and ethylidyne, Pt3![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CH3, a species that forms via ethylene dehydrogenation, are also strongly adsorbed on the surface). The probability for ethane formation decreases noticeably in this regime, and the reaction mechanism switches to the stepwise hydrogen incorporation sequence proposed by Horiuti and Polanyi many years ago. Finally, for reaction mixtures with more than 1% of ethylene, the ethylidyne surface layer reaches coverages close to saturation and controls the HD and ethane formation kinetics via site blocking; this latter regime is the one operational under most typical catalytic runs. It was also shown that the relative importance of the reversibly adsorbed ethylene and irreversibly adsorbed ethylidyne species in the reaction kinetics depends on surface temperature, and that the ethylidyne layer can be removed at temperatures around 500 K to restore the full catalytic activity of the clean Pt surface.
C–CH3, a species that forms via ethylene dehydrogenation, are also strongly adsorbed on the surface). The probability for ethane formation decreases noticeably in this regime, and the reaction mechanism switches to the stepwise hydrogen incorporation sequence proposed by Horiuti and Polanyi many years ago. Finally, for reaction mixtures with more than 1% of ethylene, the ethylidyne surface layer reaches coverages close to saturation and controls the HD and ethane formation kinetics via site blocking; this latter regime is the one operational under most typical catalytic runs. It was also shown that the relative importance of the reversibly adsorbed ethylene and irreversibly adsorbed ethylidyne species in the reaction kinetics depends on surface temperature, and that the ethylidyne layer can be removed at temperatures around 500 K to restore the full catalytic activity of the clean Pt surface.
- This article is part of the themed collection: Catalytic reactivity of surfaces: in recognition of François Gault
 Please wait while we load your content...
Please wait while we load your content...

