
Selective hydrogenation of acetylene over Cu(211), Ag(211) and Au(211): Horiuti–Polanyi mechanism vs. non-Horiuti–Polanyi mechanism†

Abstract
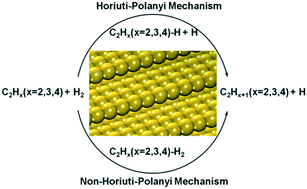
Two hydrogenation mechanisms, namely the Horiuti–Polanyi and non-Horiuti–Polanyi mechanisms, are examined and compared for acetylene hydrogenation to ethylene over Cu(211), Ag(211) and Au(211) using density functional theory (DFT) calculations. In the Horiuti–Polanyi mechanism, hydrogen molecules dissociate first followed by the sequential addition of hydrogen atoms to the hydrocarbon, whilst in the non-Horiuti–Polanyi mechanism, hydrogen molecules react with the hydrocarbon directly. It is found that the Horiuti–Polanyi mechanism is favoured on Cu(211) for the hydrogenation reactions of acetylene to ethylene, whilst the non-Horiuti–Polanyi mechanism is favoured for the reactions over Ag(211). In contrast, on Au(211) the hydrogenation of C2H2 and C2H3 follows the Horiuti–Polanyi mechanism, but the hydrogenation of C2H4 follows the non-Horiuti–Polanyi mechanism. Further analyses suggest that the non-Horiuti–Polanyi mechanism is favoured when the reactants weakly adsorb while strong adsorption gives rise to the Horiuti–Polanyi mechanism, which is consistent with the observations reported in our previous work. From the energy profiles obtained, the activity and selectivity of the hydrogenation reactions are also quantitatively estimated and compared.
 Please wait while we load your content...
Please wait while we load your content...

