
Investigating gas sorption in an rht-metal–organic framework with 1,2,3-triazole groups†
 ‡a
and
Brian
Space
*a
‡a
and
Brian
Space
*a
Abstract
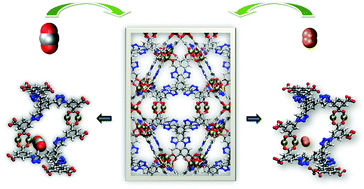
Simulations of CO2 and H2 sorption were performed in an rht-metal–organic framework (MOF) that consists of Cu2+ ions coordinated to 5,5′,5′′-(4,4′,4′′-(benzene-1,3,5-triyl)tris(1H-1,2,3-triazole-4,1-diyl))triisophthalate (BTTI) linkers; it is referred to as Cu–BTTI herein. This MOF was previously synthesized and reported by three different experimental groups [Zhao et al., Sci. Rep., 2013, 3, 1149; Schröder et al., Chem. Sci., 2013, 4, 1731–1736; Hupp et al., Energy Environ. Sci., 2013, 6, 1158–1163]. This MOF is notable for the presence of open-metal sites and nitrogen-rich regions through the copper paddlewheel ([Cu2(O2CR)4]) clusters and 1,2,3-triazole groups, respectively, which allows this material to display remarkable CO2 and H2 sorption properties. All three groups report distinct experimental and theoretical gas sorption results for the MOF. In contrast to the force fields utilized in the aforementioned studies, our simulations include explicit many-body polarization interactions, which was important to reproduce sorption onto the open-metal sites. Simulations using polarizable potentials for the MOF and sorbates generated sorption isotherms and isosteric heat of adsorption (Qst) values that are outstanding agreement with the corresponding experimental data for all three groups; this is in contrast to the theoretical results presented in the respective original references. The simulations carried out in the previous studies often looked reasonable but they missed a key feature of the sorption process that lead to unreliable results. Analysis of the radial distribution function (g(r)) about the open-metal sites and examination of the modeled structure reveal that the CO2 and H2 molecules prefer to sorb onto two unique types of Cu2+ ions that exhibit the highest partial positive charges. Sorption was also observed within the corners of the truncated tetrahedral (T-Td) cages and onto the 1,2,3-triazole groups of the linkers for both sorbates. Overall, this study demonstrates how utilizing a classical polarizable force field led to the reproduction of experimental observables and allowed for an accurate description of the sorption mechanism in this MOF that is an important member of the rht-MOF family.
 Please wait while we load your content...
Please wait while we load your content...

