
Theoretical study on geometric, electronic and catalytic performances of Fe dopant pairs in graphene†
 ‡*a
Yaqiang
Ma,
b
‡*a
Yaqiang
Ma,
b
Abstract
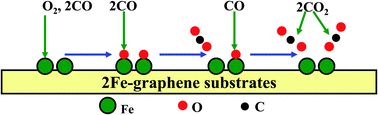
The formation geometries, electronic structures and catalytic properties of monovacancy and divacancy graphene sheets with two embedded Fe dopants (2Fe-MG and 2Fe-DG) have been systematically investigated using the first-principles calculations. It was found that the configuration of 2Fe-DG is slightly more stable than that of 2Fe-MG sheets and the two doped Fe atoms in graphene (2Fe-graphene) as active sites could regulate the stability of gas molecules. In addition, the adsorption of O2 and CO molecules could modulate the electronic and magnetic properties of 2Fe-graphene systems. Moreover, the adsorption behaviors of reactants could determine the reaction pathway and energy barrier of the catalytic oxidation of CO. On the 2Fe-graphene substrates, the adsorptive decomposition of O2 molecules (<0.20 eV) and the subsequent Eley–Rideal (ER) reaction (2Oads + 2CO → CO2) (<0.60 eV) have low energy barriers. In comparison, the CO3 complex is quite stable and its formation needs to overcome a higher energy barrier (>0.90 eV). Hence, the dissociation of O2 as an initial step is an energetically more favored process. These results provide valuable guidance for the design of functionalized graphene-based devices.
 Please wait while we load your content...
Please wait while we load your content...

