
First principles studies of CO2 and O2 chemisorption on La2O3 surfaces†
 *ac
and
*ac
and
Abstract
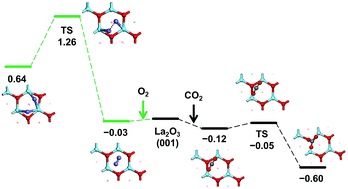
Periodic density functional theory calculations were performed to study the surface structures and stabilities of the La2O3 catalyst in CO2 and O2 environments, relevant to the conditions of the oxidative coupling of methane (OCM) reaction. Thermodynamic stabilities of the clean surfaces were predicted to follow the order of (001) ≥ (011) ≫ (110) > (111) > (101) > (100), with their direct band gaps at the Γ point following the similar order of (001) > (011) > (110) > (111) > (100) > (101). Hubbard U corrections to the La 4f and 5d orbitals do not qualitatively change the predictions of surface energies and band gaps. For the most stable (001) surface, CO2 chemisorption to form carbonate species is exothermic by −0.60 eV with a negligible energy barrier of 0.07 eV, whereas O2 chemisorption to form peroxide species is endothermic by 0.64 eV with a considerable energy barrier of 1.29 eV. For the slightly less stable (011) surface, both CO2 and O2 chemisorption can occur at different surface sites, and the same applies to the other studied surfaces. Dissociation temperatures of surface carbonate species range from 300 to 1000 K at pCO2 of 1 bar, which follow the order of (101) ≈ (110) > (111) ≈ (100) ≈ (011) ≫ (001), showing their strong sensitivity to the surface structure. Dissociation temperatures of surface peroxide species are mostly lower than the room temperature except for those of the (011) and (111) surfaces, although the significant kinetic barriers predicted should prevent their facile dissociation. Insights into the temperature-programmed desorption experiments and the methane reactivity of La2O3 in the OCM reaction were also given based on the results of our calculations.
 Please wait while we load your content...
Please wait while we load your content...

