
Catechol–cation adhesion on silica surfaces: molecular dynamics simulations†

Abstract
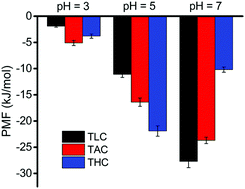
Understanding the interaction mechanism between catechol−cation and inorganic surfaces is vital for controlling the interfacial adhesion behavior. In this work, molecular dynamics simulations are employed to study the adhesion of siderophore analogues (Tren-Lys-Cam, Tren-Arg-Cam and Tren-His-Cam) on silica surfaces with different degrees of ionization and the effects of cationic amino acids and ionic strength on adhesion are discussed. Simulation results indicate that adhesion of catechol–cation onto the ionized silica surface is dominated by electrostatic interactions. At different degrees of ionization, the rank of the adhesions of three siderophore analogues on silica is different. Further analysis shows that the amino acid terminus has a large influence on the adhesion process, especially histidine adhesion on negatively charged surfaces. Tren-Lys-Cam (TLC) has a larger adhesion free energy than Tren-Arg-Cam (TAC) at a higher degree of ionization (18%); both the bulkier structure and delocalized charge of Arg decreased the cation's electrostatic interaction with the charged silica. In addition, the adhesion free energy on ionized silica surfaces decreased with increasing ionic strength of aqueous solutions. A linear correlation between the potential of mean force obtained from umbrella sampling and the rupture force via steered molecular dynamics simulations for siderophore analogue adhesion on silica surfaces is also found. This work may provide some guidance for developing the next generation underwater adhesives.
 Please wait while we load your content...
Please wait while we load your content...

