
Molecular simulations of palladium catalysed hydrodeoxygenation of 2-hydroxybenzaldehyde using density functional theory†

Abstract
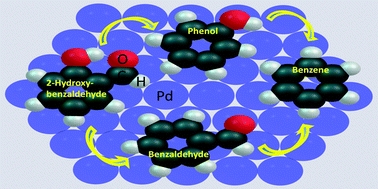
The catalytic conversion of 2-hydroxybenzaldehyde (2-HB) is carried out numerically over a Pd(111) surface using density functional theory. The palladium catalyst surface is designed using a 12 atom monolayer and verified with the adsorption of phenol, benzene, anisole, guaiacol, and vanillin; it is found that the adsorption energies along with the adsorption configurations of phenol and benzene are in excellent agreement with the literature. The conversion of 2-HB over the Pd(111) catalyst surface is performed using four reaction schemes: (i) dehydrogenation of the formyl group followed by elimination of CO and association of hydrogen with 2-hydroxyphenyl to produce phenol, (ii) direct elimination of CHO from 2-HB followed by elimination of hydrogen from adsorbed CHO and association of hydrogen with 2-hydroxyphenyl to produce phenol, (iii) direct dehydroxylation of 2-HB followed by association of a hydrogen atom with 2-formylphenyl to produce benzaldehyde, and (iv) dehydrogenation of the hydroxyl group of 2-HB followed by elimination of an oxygen atom and association of a hydrogen atom with 2-formylphenyl to produce benzaldehyde. Along with the reaction mechanisms and their barrier heights, all reaction steps are considered for kinetic modelling in the temperature range 498–698 K with 50 K intervals. The rate constants, pre-exponential factors, and equilibrium constants of all elementary reaction steps are evaluated for each temperature. Kinetic analyses of the catalytic conversion of 2-HB over the Pd(111) surface suggests the production of phenol as an intermediate, instead of benzaldehyde, via dehydrogenation of the formyl group of 2-HB as a first elementary reaction step because of its low activation barrier and the high rate constant of the rate controlling step. Furthermore, the equilibrium constants of the rate controlling step in the production of phenol from 2-HB over the Pd(111) surface report a major fraction of the product in the product mixture even at a low temperature of 498 K.
 Please wait while we load your content...
Please wait while we load your content...

