
Reaction kinetics of hydrogen abstraction from polycyclic aromatic hydrocarbons by H atoms†
 *ab
*ab
Abstract
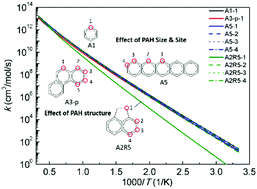
Hydrogen abstraction reactions of polycyclic aromatic hydrocarbons (PAH) by H atoms play a very important role in both PAH and soot formation processes. However, large discrepancies up to a few orders of magnitude exist among the literature rate constant values. To increase the reliability of the computed rate constants, it is critical to obtain highly accurate potential energy surfaces. For this purpose, we have investigated the energetics of hydrogen abstraction from benzene and naphthalene using both high level-of-theory quantum chemistry methods and a series of density functional theory (DFT) methods, among which M06-2X/6-311g(d,p) has the best performance with a mean unsigned deviation from the CCSD(T)/CBS calculations of 1.0 kcal mol−1 for barrier heights and reaction energies. Thus, M06-2X/6-311g(d,p) has then been applied to compute the potential energy surfaces of the hydrogen abstraction reactions of a series of larger PAH. Based on the quantum chemistry calculations, rate constants are computed using the canonical transition state theory. The effects of the PAH size, structure, and reaction site on the energetics and rate constants are examined systematically. Finally, the hydrogen abstraction rate constants for application in PAH and soot surface chemistry models are recommended.
 Please wait while we load your content...
Please wait while we load your content...

