
Fast and accurate prediction of proton affinities: revisiting the extended Koopmans' theorem for protons†
 *a
*a
Abstract
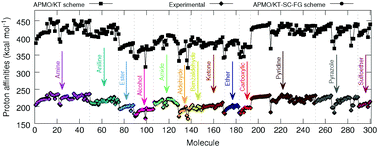
In this work we propose schemes based on the extended Koopmans' theorem for quantum nuclei (eKT), in the framework of the any particle molecular orbital approach (APMO/KT), for the quantitative prediction of gas phase proton affinities (PAs). The performance of these schemes has been tested on a set of 300 organic molecules containing diverse functional groups. The APMO/KT scheme scaled by functional group (APMO/KT-SC-FG) displays an overall mean absolute error of 1.1 kcal mol−1 with respect to experimental data. Its performance in PA calculations is similar to that of post-Hartree–Fock composite methods or that of the APMO second order proton propagator (APMO/PP2) approach. The APMO/KT-SC-FG scheme is also employed to predict PAs of polyfunctional molecules such as the Nerve Agent VX and the 20 common α-amino acids, finding excellent agreement with available theoretical and/or experimental data. The accuracy of the predictions demonstrates that the APMO/KT-SC-FG scheme is a low-cost alternative to adiabatic methods for the calculation of accurate PAs. One of the most appealing features of the APMO/KT-SC-FG scheme, is that PAs can be derived from one single-point APMO Hartree–Fock calculation.
 Please wait while we load your content...
Please wait while we load your content...

