
Density functional theory study on the metal–support interaction between a Au9 cluster and an anatase TiO2(001) surface

Abstract
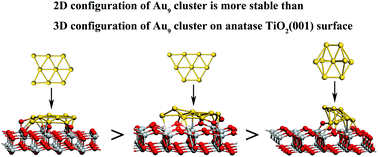
Noble metals supported on TiO2 surfaces have shown extraordinary photocatalytic properties in many important processes such as hydrogenation, water splitting, degradation of hazards, and so on. Using density functional theory calculations, this work has systematically investigated the microstructure and electronic structure of three different Au9 isomers loaded on anatase TiO2(001) surface. The calculated results show that the interaction between the Au9 cluster and the TiO2 support is closely related to the adsorption site and the stability of the Au9 cluster in the gas phase. The adsorption energy of the 2D configuration is larger than that of the 3D configuration of the Au9 cluster, owing to the stronger interactions between more adsorption sites. The stable adsorption site for Au9 clusters deposited on the anatase TiO2(001) surface tends to be the O2c–O2c hollow site. The presentation of the MIGS of the Au9 cluster, the disappearance of surface states of the TiO2(001) surface, and the shifting of the Fermi level from the top of the valence band to the bottom of the conduction band suggest strong interactions between the Au9 clusters and the TiO2(001) surface. Importantly, the electron transfer from the Au9 clusters to the TiO2 support occurs mainly through Au–O2c interactions, which are mainly localized at the contact layer of the Au9 clusters. These conclusions are useful to understand various physical and chemical properties of noble metal clusters loaded onto an oxide surface, and helpful to design novel metal/semiconductor functional composite materials and devices.
 Please wait while we load your content...
Please wait while we load your content...

