
Dissociative adsorption dynamics of nitrogen on a Fe(111) surface
 *ab
*ab
Abstract
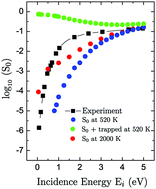
We study the dissociative adsorption dynamics of N2 on clean bcc Fe(111) surfaces. We base our theoretical analysis on a multidimensional potential energy surface built from density functional theory. The dissociative sticking probability is computed by means of quasi-classical trajectory calculations. For normal incidence and impact energies of the order of a few eV, our theoretical results agree well with existing experimental values. For these energies, the dynamics of the dissociated molecules shows that dissociation is a direct process that follows narrow paths in the multidimensional space. For lower energies of the beam, this direct process is not enough to explain the measured values. A better agreement with the experiment is obtained if we increase the surface temperature to promote the transfer to dissociation of molecules previously trapped. Most of the molecules dissociate very close to the Fe(111) third layer atoms and with an orientation parallel to the surface. A comparison between the dissociation of N2 on Fe(111) and Fe(110) highlights the role of the different energy barriers in both surfaces.
 Please wait while we load your content...
Please wait while we load your content...

