
Quantum chemical and molecular dynamics modelling of hydroxylated polybrominated diphenyl ethers†

Abstract
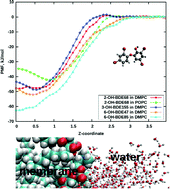
A series of 19 hydroxylated polybrominated diphenyl ethers (OH-PBDEs) have been studied using density functional theory (DFT) and molecular dynamics simulations with the purpose of investigating eventual correlations between their physicochemical properties and toxic action. Dissociation constants (pKa), solvation free energies and octanol–water partition coefficients (log P) have been computed. Additionally, metadynamics simulations of OH-PBDEs passing through a lipid bilayer have been carried out for four OH-PBDE species. No correlations between computed pKa values and toxicity data have been found. Medium correlations were found between partition coefficients and the ability of OH-PBDEs to alter membrane potential in cell cultures, which is attributed to higher uptake of molecules with larger log P parameters. It was also demonstrated that in lipid bilayers, OH-PBDE molecules differ in their orientational distributions and can adopt different conformations which can affect the uptake of these molecules and influence the pathways of their toxic action.
 Please wait while we load your content...
Please wait while we load your content...

