
Molecular dynamics simulations of pyrrolidinium and imidazolium ionic liquids at graphene interfaces†

Abstract
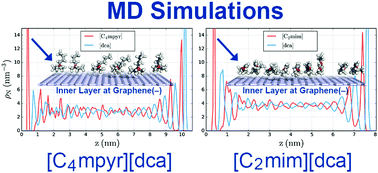
Understanding the electrode–electrolyte interface is essential in the battery research as the ion transport and ion structures at the interface most likely affect the performance of a battery. Here we investigate interfacial structures of three ionic liquids: 1-ethyl-3-methylimidazolium dicyanamide ([C2mim][dca]), 1-butyl-3-methylimidazolium dicyanamide ([C4mim][dca]) and N-butyl-N-methylpyrrolidinium dicyanamide ([C4myr][dca]) at a charged and uncharged graphene interface using molecular dynamics simulations. We find that these ionic liquids (ILs) behave differently both in the bulk phase and near a graphene interface and we find that this difference is apparent in all types of analyses performed here. First, a partial density analysis in the direction perpendicular to the surface of the electrodes, which, in the cases near a negatively charged graphene, reveals that the pyrrolidinium system is generally more layered than the imidazolium systems. Second, a 2D topographic structure analysis of the IL species in the inner layer near a negatively charged graphene surface, which reveals that the pyrrolidinium system exhibits a quasi-hexagonal surface configuration of the cations, while the imidazolium systems show linearly arranged groups of cations. Third, a 3D orientation-preference analysis of cation rings near the negative graphene electrode, which shows that the pyrrolidinium rings prefer to lie parallel to the electrode surface while the imidazolium rings prefer to stand on the electrode surface at high tilt angles. Extending the imidazolium alkyl chain was found to reduce the number of imidazoliums that can link up into linearly arranged groups in the inner layer 2D structures. Our results support earlier experimental findings and indicate that the interfacial nanostructures may have a significant influence on the electrochemical performance of IL-based batteries.
 Please wait while we load your content...
Please wait while we load your content...

