
Density functional theory calculations for the band gap and formation energy of Pr4−xCaxSi12O3+xN18−x; a highly disordered compound with low symmetry and a large cell size†

Abstract
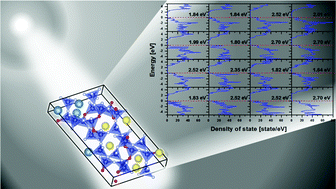
A novel oxynitride compound, Pr4−xCaxSi12O3+xN18−x, synthesized using a solid-state route has been characterized as a monoclinic structure in the C2 space group using Rietveld refinement on synchrotron powder X-ray diffraction data. The crystal structure of this compound was disordered due to the random distribution of Ca/Pr and N/O ions at various Wyckoff sites. A pragmatic approach for an ab initio calculation based on density function theory (DFT) for this disordered compound has been implemented to calculate an acceptable value of the band gap and formation energy. In general, for the DFT calculation of a disordered compound, a sufficiently large super cell and infinite variety of ensemble configurations is adopted to simulate the random distribution of ions; however, such an approach is time consuming and cost ineffective. Even a single unit cell model gave rise to 43 008 independent configurations as an input model for the DFT calculations. Since it was nearly impossible to calculate the formation energy and the band gap energy for all 43 008 configurations, an elitist non-dominated sorting genetic algorithm (NSGA-II) was employed to find the plausible configurations. In the NSGA-II, all 43 008 configurations were mathematically treated as genomes and the calculated band gap and the formation energy as the objective (fitness) function. Generalized gradient approximation (GGA) was first employed in the preliminary screening using NSGA-II, and thereafter a hybrid functional calculation (HSE06) was executed only for the most plausible GGA-relaxed configurations with lower formation and higher band gap energies. The final band gap energy (3.62 eV) obtained after averaging over the selected configurations, resembles closely the experimental band gap value (4.11 eV).
 Please wait while we load your content...
Please wait while we load your content...

