
An adaptive bias – hybrid MD/kMC algorithm for protein folding and aggregation†
 *a
and
Joan-Emma
Shea
b
*a
and
Joan-Emma
Shea
b
Abstract
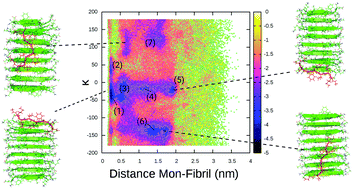
In this paper, we present a novel hybrid Molecular Dynamics/kinetic Monte Carlo (MD/kMC) algorithm and apply it to protein folding and aggregation in explicit solvent. The new algorithm uses a dynamical definition of biases throughout the MD component of the simulation, normalized in relation to the unbiased forces. The algorithm guarantees sampling of the underlying ensemble in dependency of one average linear coupling factor 〈α〉τ. We test the validity of the kinetics in simulations of dialanine and compare dihedral transition kinetics with long-time MD-simulations. We find that for low 〈α〉τ values, kinetics are in good quantitative agreement. In folding simulations of TrpCage and TrpZip4 in explicit solvent, we also find good quantitative agreement with experimental results and prior MD/kMC simulations. Finally, we apply our algorithm to study growth of the Alzheimer Amyloid Aβ 16–22 fibril by monomer addition. We observe two possible binding modes, one at the extremity of the fibril (elongation) and one on the surface of the fibril (lateral growth), on timescales ranging from ns to 8 μs.
 Please wait while we load your content...
Please wait while we load your content...

