
Modelling aqueous solubility of sodium chloride in clays at thermodynamic conditions of hydraulic fracturing by molecular simulations†

Abstract
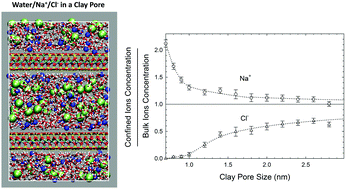
To address the high salinity of flow-back water during hydraulic fracturing, we have studied the equilibrium partitioning of NaCl and water between the bulk phase and clay pores. In shale rocks, such a partitioning can occur between fractures with a bulk-like phase and clay pores. We use an advanced Grand Canonical Monte Carlo (GCMC) technique based on fractional exchanges of dissolved ions and water molecules. We consider a typical shale gas reservoir condition of a temperature of 365 K and pressure of 275 bar, and we represent clay pores by pyrophyllite and Na-montmorillonite slits of a width ranging from about 7 to 28 Å, covering clay pores from dry clay to clay pores with a bulk-like layer in the middle of the pore. We employ the Joung–Cheatham model for ions, SPC/E model for water and CLAYFF for the clay pores. We first determine the chemical potentials for NaCl and water in the bulk phase using Osmotic Ensemble Monte Carlo simulations. The chemical potentials are then used in GCMC to simulate the adsorption of ions and water molecules in the clay pores, and in turn to predict the salt solubility in confined solutions. Besides the thermodynamic properties, we evaluate the structure and in-plane diffusion of the adsorbed fluids, and ion conductivities.
 Please wait while we load your content...
Please wait while we load your content...

