
Chemodynamics of metal ion complexation by charged nanoparticles: a dimensionless rationale for soft, core–shell and hard particle types

Abstract
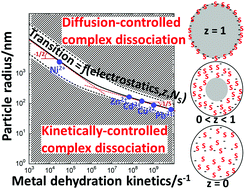
Soft nanoparticulate complexants are defined by a spatial confinement of reactive sites and electric charges inside their 3D body. In turn, their reactivity with metal ions differs significantly from that of simple molecular ligands. A revisited form of the Eigen mechanism recently elucidated the processes leading to metal/soft particle pair formation. Depending on e.g. particle size and metal ion nature, chemodynamics of nanoparticulate metal complexes is controlled by metal conductive diffusion to/from the particles, by intraparticulate complex formation/dissociation kinetics, or by both. In this study, a formalism is elaborated to achieve a comprehensive and systematic identification of the rate-limiting step governing the overall formation and dissociation of nanoparticulate metal complexes. The theory covers the different types of spherical particulate complexants, i.e. 3D soft/permeable and core–shell particles, and hard particles with reactive sites at the surface. The nature of the rate-limiting step is formulated by a dynamical criterion involving a power law function of the ratio between particle radius and an intraparticulate reaction layer thickness defined by the key electrostatic, diffusional and kinetic components of metal complex formation/dissociation. The analysis clarifies the intertwined contributions of particle properties (size, soft or hard type, charge, density or number of reactive sites) and aqueous metal ion dehydration kinetics in defining the chemodynamic behavior of nanoparticulate metal complexes. For that purpose, fully parameterized chemodynamic portraits involving the defining features of particulate ligand and metal ion as well as the physicochemical conditions in the local intraparticulate environment, are constructed and thoroughly discussed under conditions of practical interest.
- This article is part of the themed collection: 2017 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

